Model Status
This CellML version of the model has been checked in COR and OpenCell and the model runs to reproduce the results as shown in figure 3 of the published paper (ie. k69=0). The units have been checked and are consistent. This CellML description is deterministic, unlike the stochastic model created and preferred by the authors. Time courses produced are therefore similar to, but not exactly the same as the figures in the paper; the graphs produced by this CellML description have smooth curves since random fluctuations of the intracellular preocesses are not taken into account.
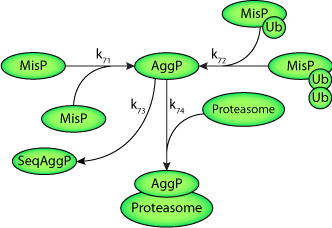
Model Structure
ABSTRACT: The ubiquitin-proteasome system is responsible for homeostatic degradation of intact
protein substrates as well as the elimination of damaged or misfolded proteins that might otherwise
aggregate. During ageing there is a decline in proteasome activity and an increase in aggregated proteins.
Many neurodegenerative diseases are characterised by the presence of distinctive ubiquitin-positive
inclusion bodies in affected regions of the brain. These inclusions consist of insoluble, unfolded,
ubiquitinated polypeptides that fail to be targeted and degraded by the proteasome. We are using a
systems biology approach to try and determine the primary event in the decline in proteolytic capacity
with age and whether there is in fact a vicious cycle of inhibition, with accumulating aggregates further
inhibiting proteolysis, prompting accumulation of aggregates and so on.The model can be used to predict the
effects of different experimental procedures such as inhibition of the proteasome or shutting down the
enzyme cascade responsible for ubiquitin conjugation. The model output shows good agreement with experimental data under a number of different
conditions. However, our model predicts that monomeric ubiquitin pools are always depleted under
conditions of proteasome inhibition, whereas experimental data show that monomeric pools were
depleted in IMR-90 cells but not in ts20 cells, suggesting that cell lines vary in their ability to replenish
ubiquitin pools and there is the need to incorporate ubiquitin turnover into the model. Sensitivity analysis
of the model revealed which parameters have an important effect on protein turnover and aggregation
kinetics.
The original paper reference is cited below:
An in silico model of the ubiquitin-proteasome system that incorporates normal homeostasis and age-related decline, Carole J. Proctor, Maria Tsirigotis and Douglas A. Gray, 2007,BMC Systems Biology, 1:17. PubMed ID: 17408507